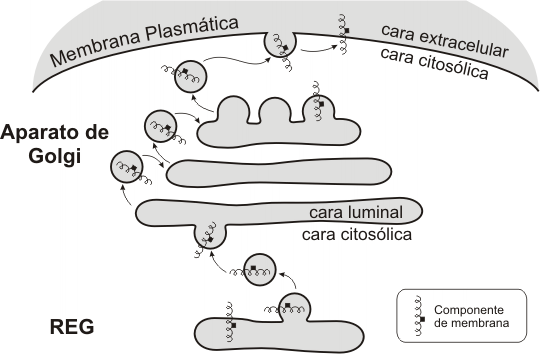
Funciones del retículo endoplasmático liso
a) Síntesis de lípidos. En las membranas del REL se sitúan las enzimas responsables de la síntesis de la mayor parte de los lípidos celulares: triglicéridos, fosfoglicéridos, ceramidas y esteroides. Los precursores para la síntesis provienen del citosol, hacia el cual se orientan los sitios activos de las respectivas enzimas. Por lo tanto, los lípidos recién sintetizados quedan incorporados en la monocapa citosólica del REL. Sin embargo, gracias a la participación de las flipasas del retículo, se logra el movimiento hacia la monocapa luminal de los lípidos correspondientes, asegurándose de esta forma la asimetría entre ambas capas, que será mantenida de aquí en más.
b) El REL en las células musculares. El REL actúa como reservorio de calcio, el cual –frente a la llegada de un estímulo - es liberado al citosol, donde dispara una respuesta específica. Esta función es particularmente importante en las células musculares. Allí el REL, que toma el nombre de retículo sarcoplásmico, adopta una conformación muy especializada. El calcio es liberado frente al impulso nervioso desencadenado por la acetil colina en la unión neuromuscular, y una vez en el citosol participa en la contracción muscular. Cuando retorna al REL, por la acción de una bomba de calcio, se produce la miorrelajación.
c) El REL en las células hepáticas. Está involucrado en dos funciones: detoxificación y glucogenólisis. La detoxificación consiste en la transformación de metabolitos y drogas en compuestos hidrosolubles que puedan ser excretados por orina.
La glucogenólisis (degradación del glucógeno) tiene lugar en el citosol, donde los gránulos de glucógeno se encuentran en íntima relación con el REL. El producto de la glucogenólisis, la glucosa 6-fosfato (glucosa 6-P), es atacada entonces por la glucosa 6-fosfatasa, enzima de la membranas del retículo. Ésta cataliza la hidrólisis del grupo fosfato, permitiendo así que la glucosa atraviese la membrana celular hacia el torrente circulatorio. La glucosa 6-fosfatasa no se expresa en las células musculares, razón por la cual el glucógeno muscular no contribuye a la mantención de la glucemia
Funciones del retículo endoplasmático granular
a) Síntesis de proteínas. Todas las proteínas sintetizadas en la célula (excepto las codificadas por ADN de mitocondria y cloroplasto) son iniciadas por ribosomas libres del citosol. Muchas de ellas, las proteínas nucleares, las citosólicas y las que están destinadas a cloroplastos, mitocondrias o peroxisomas, concluyen su síntesis en dichos ribosomas para luego dirigirse, por el citosol, hacia sus compartimentos diana. Otras, en cambio, como las proteínas integrales de membrana, las de secreción y las enzimas lisosomales, terminan su síntesis en el REG. ¿Cómo se dirige la síntesis hacia uno de estos dos ramales? ¿Existen diferentes poblaciones de ribosomas? ¿Dónde radica la señal que conduce a determinadas proteínas hacia el REG
Síntesis de proteínas en el REG. Hipótesis de la señal
La respuesta a estos interrogantes fue proporcionada por Blobel y Sabatini, en el año 1971, cuando propusieron su hipótesis de la señal, ampliamente corroborada después. Las proteínas que se sintetizan en el REG tienen en su extremo aminoterminal una seguidilla de aproximadamente treinta aminoácidos cuyos radicales son predominantemente hidrófobos. Este primer fragmento de las proteínas recibe el nombre de péptido señal o péptido guía. No aparece péptido guía en las proteínas del citosol, núcleo, mitocondria, cloroplasto ni peroxisoma. Cuando el péptido guía está presente, es reconocido por la PRS (partícula de reconocimiento de la señal) situada en el citosol. La PRS interactúa con el péptido señal y detiene la síntesis temporariamente. Entonces el ribosoma se une a las membranas del REG. Recordemos que allí se ubican las riboforinas (receptores de ribosomas). También hay receptores para la PRS. Una vez que el ribosoma se adhiere a las membranas reticulares, entonces el péptido guía ingresa en un canal transmembranar, la PRS se separa y la traducción se reanuda. A medida que la proteína crece, se vuelca hacia el lumen del REG: la síntesis proteica y la translocación a través de la membrana son simultáneas (cotraslación).
).
Sïntesis de proteínas en el REG. Cotraslación
Las proteínas que carecen de péptido señal no son reconocidas por la PRS; por este motivo no se dirigen hacia el sistema de endomembranas y su síntesis se completa en el citosol. Muchas de ellas atraviesan otras membranas con posterioridad (postraslación) para alcanzar su localización definitiva. Se han encontrado otras secuencias aminoacídicas, distintas del péptido señal, que actúan como marcas para dirigirlas a sus respectivos destinos.
Las proteínas sintetizadas en el REG pueden dividirse en dos grandes grupos: membranares y luminales o solubles. Las membranares permanecen incluidas en la membrana, en algunos casos ligadas a ella mediante el péptido señal; en otras, secuencias de aminoácidos internas a la cadena funcionan como péptidos de anclaje, deteniendo la translocación de la proteína por el canal. Según la cantidad de secuencias de anclaje que presentan, hay proteínas de paso único o proteínas multipaso. Las proteínas intrínsecas insertas en la membrana a nivel del REG se retienen como componentes de este organoide o son transportadas en vesículas, formando parte del “envase”, hasta incorporarse a otras membranas del sistema o a la propia membrana plasmática.
- Inserción de proteínas integrales en la membrana del
Inserción de proteínas integrales de multiple paso en la membrana del reg
Funciones del aparato de Golgi
Endosoma temprano y tardío
Las proteínas solubles no conservan el péptido señal ni poseen otros péptidos de anclaje. Cuando el péptido señal es escindido de la cadena (en este corte actúa una peptidasa señal ubicada en la cara luminal de las cisternas), ésta pierde contacto con la membrana y se vuelca por completo al lumen. Si las proteínas solubles no son residentes del REG, entonces siguen su ruta, en este caso como contenido de las vesículas transportadoras. Podemos citar en este grupo a las proteínas de secreción y a las hidrolasas lisosomales.
Glicosilación. La mayor parte de las proteínas sintetizadas en el REG incorporan cadenas glucídicas a su paso por el mismo. La presencia en la cadena polipeptídica de la secuencia de aminoácidos asparagina–x-serina o asparagina–x–treonina (x es otro aminoácido cualquiera), señal de glicosilación, marca el sitio donde se unirá el glúcido. Todas las glucoproteínas sintetizadas en el REG reciben el mismo oligosacárido: una cadena ramificada de doce unidades de monosacárido.
- Glicosilación nuclear.
Ésta se sintetiza sobre un lípido de membrana -el dolicol-fosfato -, y luego es transferida en bloque a la asparagina de la señal de glicosilación (se forma un enlace N-glicosídico). En la síntesis del oligosacárido y su posterior transferencia participan las enzimas glicosiltransferasas. El glúcido se adiciona tantas veces como aparezca en la proteína la señal de glicosilación. Mientras la glucoproteína aún se halla en el REG, enzimas glicosidasas remueven algunas unidades de monosacárido, al tiempo que distintas glicosiltransferasas añaden otras nuevas. Se produce así la diversidad de cadenas a partir del primer bloque transferido. Un núcleo del oligosacárido original, no obstante, se conserva hasta el final en todas las glucoproteínas, de allí que esta glicosilación reciba el nombre de glicosilación
Funciones del aparato de Golgi
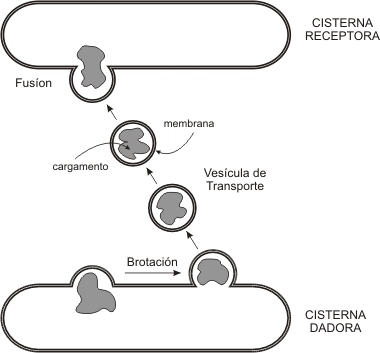
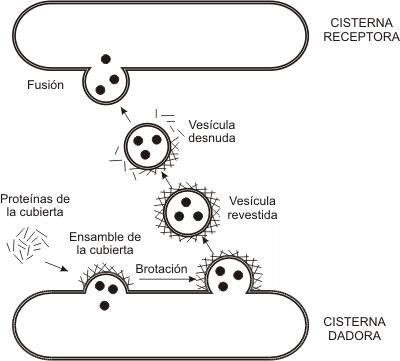
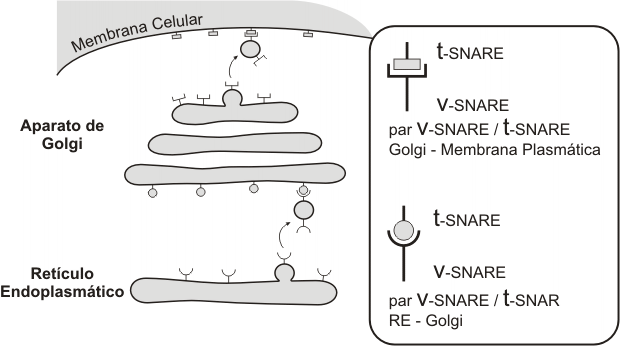
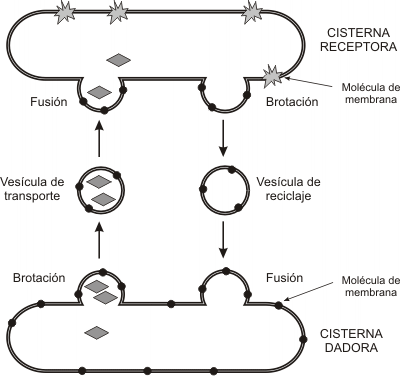
El aparato de Golgi es la estación distribuidora final del sistema. Las macromoléculas sintetizadas en REL y REG llegan a él mediante transporte vesicular y son recibidas en la cara convexa del aparato o cara de recepción, donde se encuentra una zona de transición con el RE, la red cis. Desde allí, por el mismo mecanismo, son enviadas a la cisterna cis, luego a la medial, y por último al compartimento trans del complejo de Golgi, que se corresponde con su cara cóncava. A partir de otra zona de transición, la red del trans Golgi, brotan las vesículas que contienen los productos definitivos.
El complejo de Golgi no se limita al transporte de las sustancias recibidas. En este sector, por el contrario, se da la forma final a las moléculas que ingresan. En el aparato de Golgi tienen lugar las siguientes reacciones:
El complejo de Golgi no se limita al transporte de las sustancias recibidas. En este sector, por el contrario, se da la forma final a las moléculas que ingresan. En el aparato de Golgi tienen lugar las siguientes reacciones:
Glicosilación terminal. Es la modificación secuencial, por remoción y adición de monosacáridos, de las glucoproteínas sintetizadas en el REG. También se adicionan nuevos bloques oligosacarídicos construidos por completo en el aparato de Golgi, proceso denominado O-glicosilación (el enlace entre el glúcido y el resto de aminoácido es unión O-glicosídica).
Síntesis de heteropolisacáridos. Los heteropolisacáridos constituyentes de los glicosaminoglicanos (GAG) se sintetizan en el aparato de Golgi y se unen a las proteínas provenientes del REG, ensamblando moléculas complejas como el ácido hialurónico o el condroitín-sulfato destinados a la matriz extracelular de las células animales. En las células vegetales, el aparato de Golgi sintetiza polisacáridos de la pared celular, por ejemplo hemicelulosas y pectinas.
Síntesis de glucolípidos. Se adiciona la porción glucídica a la ceramida sintetizada en el REL.
Secreción
Las vesículas que brotan de la cara trans portan los productos acabados destinados al medio extracelular. La fusión de dichas vesículas con la membrana plasmática –exocitosis- da como resultado la secreción o exportación de diversas sustancias: enzimas, hormonas, moléculas de la matriz extracelular o de la pared celular, anticuerpos y otras, según el tipo celular.
Hay dos rutas secretorias: la continua o constitutiva y la discontinua o regulada.
La secreción continua o constitutiva está presente en todos los tipos celulares. Las vesículas que siguen esta ruta se exocitan en forma continua, a medida que brotan del aparato de Golgi. Por ejemplo, se secretan por esta vía las moléculas que se incorporan a la matriz extracelular.
Secreción
La secreción regulada, en cambio, es propia de células secretoras especializadas. En estos casos, las vesículas se acumulan en el polo secretor de la célula, como gránulos de secreción, y la exocitosis se dispara sólo ante señales muy específicas. Por ejemplo, las células b de los islotes de Langerhans (en el páncreas), contiene gránulos de insulina que son exocitados en respuesta a una elevación de la glucemia. La secreción regulada requiere también un aumento de la concentración de calcio citosólico.
FORMACION DE LISOSOMAS PRIMARIOSLos lisosomas primarios son organoides derivados del sistema de endomembranas. Cada lisosoma primario es una vesícula que brota del aparato de Golgi, con un contenido de enzimas hidrolíticas (hidrolasas). Las hidrolasas son sintetizadas en el REG y viajan hasta el aparato de Golgi por transporte vesicular. Allí sufren una glicosilación terminal de la cual resultan con cadenas glucídicas ricas en manosa-6-fosfato (manosa 6-P). La manosa 6-P es el marcador molecular, la “estampilla” que dirige a las enzimas hacia la ruta de los lisosomas. Se ha estudiado una enfermedad en la cual las hidrolasas no llevan su marcador; las membranas del aparato de Golgi no las reconocen como tales y las empacan en vesículas de secreción para ser exocitadas. Quienes padecen esta enfermedad acumulan hidrolasas en el medio extracelular, mientras sus células carecen de ellas.
Lisosomas y digestión: heterofagia y autofagia
Los lisosomas primarios contienen una variedad de enzimas hidrolíticas capaces de degradar casi todas las moléculas orgánicas. Estas hidrolasas se ponen en contacto con sus sustratos cuando los lisosomas primarios se fusionan con otras vesículas. El producto de la fusión es un lisosoma secundario. Por lo tanto, la digestión de moléculas orgánicas se lleva a cabo en los lisosomas secundarios, ya que éstos contienen a la vez los sustratos y las enzimas capaces de degradarlos.
Existen diversas formas de lisosomas secundarios, según el origen de la vesícula que se fusiona con el lisosoma primario:
Fagolisosoma: se origina de la fusión del lisosoma primario con una vesícula procedente de la fagocitosis. Se encuentran, por ejemplo, en los glóbulos blancos, capaces de fagocitar partículas extrañas que luego son digeridas en estos cuerpos.
Endosoma tardío: surge al unirse los lisosomas primarios con materiales provenientes de los endosomas tempranos. Los endosomas tempranos contienen macromoléculas que ingresan por los mecanismos de endocitosis inespecífica y endocitosis mediada por receptor. Este último es utilizado por las células para incorporar, por ejemplo, las lipoproteínas de baja densidad o LDL.
Endosoma temprano y tardío
Autofagolisosoma: es el producto de la fusión entre un lisosoma primario y una vacuola autofágica. Algunos organoides citoplasmáticos son englobados en vacuolas, con membranas provistas por las cisternas del RE, para luego ser reciclados cuando estas vacuolas autofágicas se unen con los lisosomas primarios.
La digestión que tiene lugar en los lisosomas secundarios, ya se trate de una heterofagia- hidrólisis de sustancias de origen exógeno- o de una autofagia –degradación de componentes celulares- da origen a moléculas más sencillas que atraviesan la membrana lisosomal, es decir son absorbidas por el citosol para su posterior asimilación.
Lo que queda del lisosoma secundario después de la absorción es un cuerpo residual. Los cuerpos residuales contienen desechos no digeribles que en algunos casos se exocitan y en otros no, acumulándose en el citosol a medida que la célula envejece. Un ejemplo de cuerpos residuales son los gránulos de lipofuscina que se observan en células de larga vida, como las neuronas.
La activación de las hidrolasas requiere un medio más ácido que el citosol, de pH 5, que se logra por la acción de una bomba de protones situada en la membrana lisosomal. Por otra parte, la membrana de los lisosomas es impermeable a las enzimas y resistente a la acción de éstas. Ambos hechos protegen normalmente a la célula de una batería enzimática que podría degradarla. Existen, sin embargo, algunos procesos patológicos, como la artritis reumatoidea, que causan la destrucción de las membranas lisosomales, con la consecuente liberación de las enzimas y la lisis celular. En otros casos, la liberación de las hidrolasas cumple un papel fisiológico, permitiendo la reabsorción de estructuras que ya no son útiles, por ejemplo la cola de los renacuajos durante la metamorfosis.
Autofagia y Heterofagia
Peroxisomas
Los peroxisomas, organoides presentes en todas las células eucariontes, son vesículas ovoideas de aproximadamente 0,5 mm, que al igual que los lisosomas están rodeadas por una membrana simple y contienen enzimas en su interior. Esta quizá sea la única similitud, pues se originan al igual que las mitocondrias por un proceso de fisión binaria, en este caso de peroxisomas preexistentes. Las enzimas que contienen en su matriz se incorporan desde el citosol, siendo sintetizadas en ribosomas libres. Según el tipo de enzimas que posean, existen muchos tipos de peroxisomas.
La principal enzima de los peroxisomas es la catalasa, que descompone el peróxido de hidrógeno producido en el peroxisoma o el originado en otras localizaciones, como el citosol, RE y las mitocondrias. La actividad de la catalasa es la única común a todos los tipos de peroxisomas.
En el peroxisoma, se reduce el oxígeno molecular en dos pasos. En el primero una oxidasa elimina los electrones de varios sustratos, como aminoácidos o ácido úrico. En el segundo, la catalasa, convierte el peróxido de hidrógeno, formado en el primer paso en agua.
La catalasa también participa en la neutralización de los aniones superóxido, O2- (radicales libres). Estos radicales son primero eliminados con formación de H2O2 por la superóxido dismutasa, y luego la catalasa de los peroxisomas convierte al H2O2 en H2O y O2.
La catalasa también neutraliza con consumo de H2O2, sustancias tóxicas, como fenoles, formaldehído y el etanol de las bebidas alcohólicas, por eso son más numerosos en el tejido hepático y renal.
Contiene además diferentes oxidasas, como la D-aminooxidasa, urato oxidasa y las responsables de la b-oxidación de los ácidos grasos (este proceso tiene lugar principalmente en la mitocondria). Todas estas enzimas oxidan sus sustratos produciendo energía térmica en lugar de ATP.
En las células vegetales, encontramos glioxisomas, que son peroxisomas especializados en el metabolismo de los triacilgliceridos.
Las enzimas de los glioxisomas, transforman los ácidos grasos de las semillas en hidratos de carbono por la vía del glioxilato.
Los glioxisomas, también juegan un papel central en la fotorrespiración (se denomina así dicho proceso por requerir luz y O2 y liberar CO2), que tiene lugar en las hojas de las plantas verdes en los días de calor intenso y baja humedad ambiente.
En los glioxisomas, se cataliza la oxidación del glicocolato a H2O2 y glioxilato con consumo de oxígeno. Luego el H2O2 formado es descompuesto y el glioxilato es transformado en glicina, la cual ingresa al ciclo de Krebs.